“I routinely call hospitals and request their yearly antibiotic susceptibility testing data,” said Washington University in St. Louis’ Timothy Wencewicz. “The log might say, for example, that they’ve treated hundreds of patients for Acinetobacter baumanni, a bacterium brought into U.S. hospitals by soldiers wounded in the Iraq war, with 30 different antibiotics. The column listing the percentage of patients that responded to each antibiotic might say 1 percent or 2 percent. What happened to the other 99 percent of the patients? It’s frightening. You should see at least one drug that has a 100 percent response rate. But you don’t.”
Listening to Wencewicz can be unsettling. Wencewicz, PhD, assistant professor of chemistry in Arts & Sciences, has carefully studied antibiotics and antibiotic resistance, and very little of what he has learned is reassuring.
On the other hand, since he knows most of the moves and counter-moves that have been made in this deadly game, he has at least a fighting chance of discovering antibiotics that will stem the rising tide of antibiotic resistance.
Over the past decade, most of the big pharmaceutical companies have dropped their antibiotic discovery programs. No new antibiotics were being found by traditional methods and new methods — including genome sequencing, combinatorial chemistry and high-throughput screening — had yielded almost nothing.
And so the burden of replenishing the antibiotic pipeline has fallen on the shoulders of small biotech companies and university labs, said Wencewicz. He is under no illusion that a small lab can compete with big companies that can assign hundreds of biologists and chemists to a problem.
“What we can do,” he said, “is to discover new antibiotics made by plants, fungi or bacteria and learn enough about the details of how these molecules work that a big company will bite on and develop them.”
The key, he said, is to find drug candidates with entirely new scaffolds (core structures). Every major class of antibiotics in use today was discovered between the 1940s and the 1960s, called the “golden age” of antibiotic discovery. Chemists have been able to deal with successive waves of resistance by tailoring the chemical groups on the periphery of antibiotic molecules — the “decoration” — while leaving the core intact, but this strategy is failing.
New molecular scaffolds that act on different biological targets are desperately needed to manage antibiotic resistance over the long term, he said. One promising candidate, an antibiotic made by a bacterium than infects plants, caught his attention because it contains an “enchanted ring,” the beta-lactam ring that is found in penicillin and the cephalosporins, but the ring in the plant molecule acts against a different target than the traditional beta-lactam antibiotics.
Its target, glutamine synthetase, is an enzyme the microbes that cause tuberculosis (TB) use to build their cell envelope. About a third of the world’s population has latent TB (people carry the bacterium but are not yet ill). Bacterial strains resistant to at least one of the first-line drugs used against TB have been found in every country the World Health Organization has surveyed.
The strange history of antibiotics
Wencewicz comes to WUSTL from the lab of Christopher T. Walsh, PhD, at Harvard Medical School. Walsh knows so much about antibiotic discovery that he is said to have served as a kind of walking institutional memory for the companies with whom he consulted.
As one of Walsh’s last postdoctoral students, Wencewicz was given a unique parting gift: a “minus-80 freezer,” or deep cold freezer that holds DNA constructs, cell lines, bacteria — almost everything the lab generated. In effect, Walsh gave Wencewicz his lab’s long-term memory.
Last year Walsh and Wencewicz co-authored a review article in a Christopher T. Walsh honorary issue of The Journal of Antibiotics that summarized the last 50 years of antibiotic discovery. The golden age for the discovery of natural antibiotics of clinical significance lasted just 20 years, from 1940-1960, Wencewicz said.
The new miracle drugs were found in all sorts of odd places, such as the sewers of Sardinia, but many of them came from the soil. “(Pharmaceutical companies) Eli Lilly and Merck sent teams around the world to screen soil samples for new bacteria and fungi,” Wencewicz said. “They would ferment the soil microbes and screen the fermentation broths for biologically active components. That’s how almost all of the antibiotics were found.”
“Some microbes in the soil have been producing bacteria-killing compounds for millions or billions of years,” said Gautam Dantas, PhD, assistant professor of pathology and immunology in the Washington University School of Medicine in St. Louis.
“Scientists have long debated whether these compounds are produced in the soil at concentrations approaching their therapeutic doses in human medicine or at much lower concentrations, which might mean they play roles other than as weapons,” Dantas said. In the end, we know very little about soil bacteria, only about 1 in 100 of which can even be grown in culture.
But, Wencewicz said, a microbe cannot deploy these chemical weapons against the cellular processes it shares with other microbes unless it is itself immune to the chemical’s effects. So as microbes evolved genes to synthesize antibiotics, they also evolve genes to disable the antibiotics.
“Bacteria have been under immense selective pressure for millions or billions of years to develop resistance,” Dantas said. And so the soil is an immense reservoir of resistance. This is why antibiotic resistance emerges with such startling speed after new antibiotic drugs areintroduced, sometimes within a year. Pathogens just have to swap in resistance genes from the ancient reservoir of resistance in the soil microbes,” Dantas said.
By 1960, Wencewicz said, the low-hanging fruit had been picked, and it became harder to find new molecules to replace ones hobbled by resistance. The “golden age of discovery” yielded to what Wencewicz calls a “golden age of medicinal chemistry.”
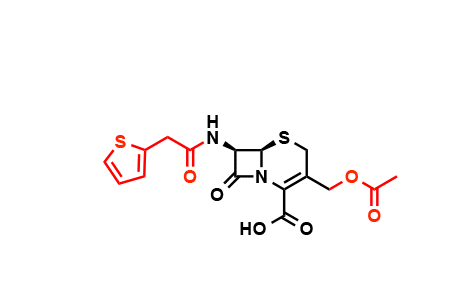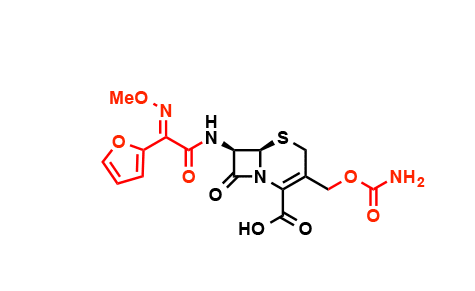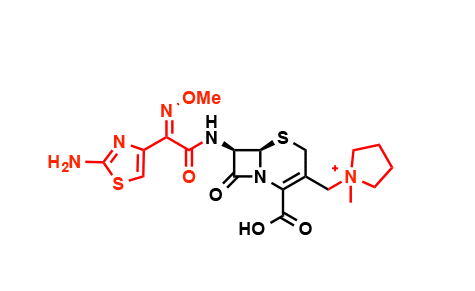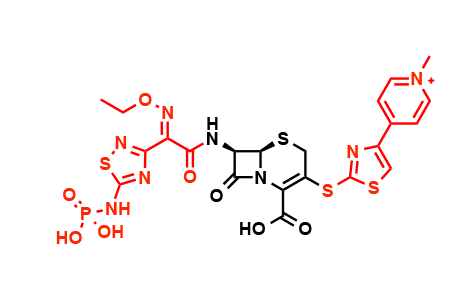
After the initial burst of discoveries, pharmaceutical companies kept one jump ahead of antibiotic resistance by altering chemical groups on the periphery of the antibiotic molecule, as shown here in the case of the cephalosporins, a strategy ultimately doomed to failure. (Credit: Wencewicz)
Chemists at the big pharmaceutical companies added or altered chemical groups on antibiotics while leaving their core intact. In this way cephalosporin became cefalotin, cefuroxime, ceflazidime, ceflepime, and cetaroline, all of which have the same core structure.
But, he said, incremental modification of these agents does not fundamentally alter their interaction with their targets and it was inevitable that this strategy would eventually fail. “You’re really just kicking the can down the road, because you’re using the same chemical scaffold and the same biological target,” he said. “It’s not a long-term management plan for antibiotic resistance.”
What happened to big pharma?
For a while, in the 1990s, huge advances in basic science and in laboratory technology promised to overleap the barriers of the past and lead to a new golden age. The two crucial developments were the ability to sequence the DNA of organisms (genomics) and souped-up synthetic chemistry, called combinatorial chemistry.
In his review, Wencewicz cites a paper by GlaxoSmithKline scientists describing an all-out attempt to use genomics and combinatorial chemistry in an antibiotic discovery program. The company spent 7 years identifying targets in the H. influenza genome that, if hit, would kill the bacterium, and then ran 70 high-throughput screens, at a cost of $1 million per screen, looking for chemical compounds that would be effective against those targets.
To everyone’s shock, they found nothing.
This demoralizing result eventually led to the abandonment of the major antibiotic discovery efforts at GlaxoSmithKline. Most other big pharmaceutical companies — Pfizer, Eli Lilly, Merck — got out as well.
A Trojan Horse
Given the flaming wrecks littering the landscape of antibiotic discovery, it took a certain amount of courage to try again. And yet people did. Wencewicz was particularly inspired by the example of pharmaceutical scientist Steven J. Brickner.
Working on his own time, Brickner, PhD, and two colleagues developed a synthetic compound into linezolid, a drug effective against the superbug methicillin-resistant S. aureus (MRSA) and other resistant Gram-positive bacteria.
Linezolid was the first member of an entirely new class of antibiotics to reach the market in 40 years.
“The development of a molecule with a new structure and new mechanism of action was a game changer,” Wencewicz said. “Brickner redefined how collaborative drug discovery works and set a new standard for bringing a drug to market.
To duplicate Brickner’s success Wencewicz has to find a compound with a new structure. “Every day there are new molecules published and patented in the literature,” he said. Most of these potential drug candidates are not pursued, and so old patent literature is a great place to look for new antibiotic candidates, Wencewicz said.
He is currently working on a molecule called tabtoxin, which is produced by a pathogen that causes wildfire disease in plants. “I was interested in this molecule because of its unusual structure,” he said. “It contains abeta-lactam, the enchanted ring that gives the penicillins their antibiotic properties but it hits a different biological target than penicillin.”
The penicillins target enzymes called transpeptidases that crosslink molecules in a bacterium’s cell wall, giving it strength. If those enzymes are inhibited the cells lose integrity and rupture.
Tabtoxin, however, acts against an enzyme called glutamine synthetase that converts glutamic acid to glutamine, an essential amino acid that plays many biochemical roles. When the enzyme is blocked by the drug, two things happen: toxic levels of ammonia build up in the cell, and the cell is starved of an essential amino acid.
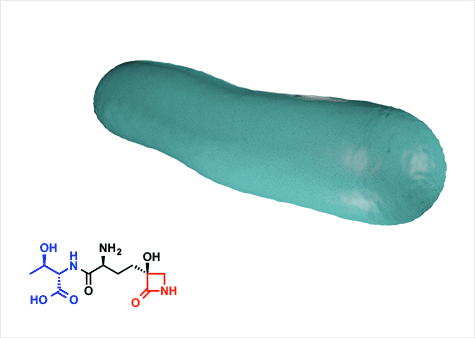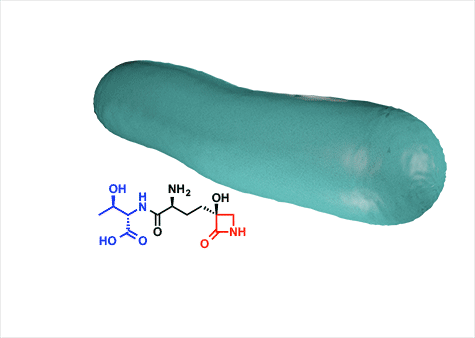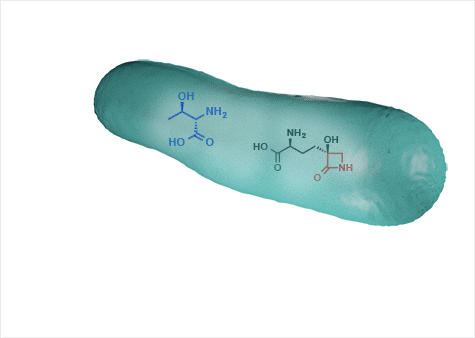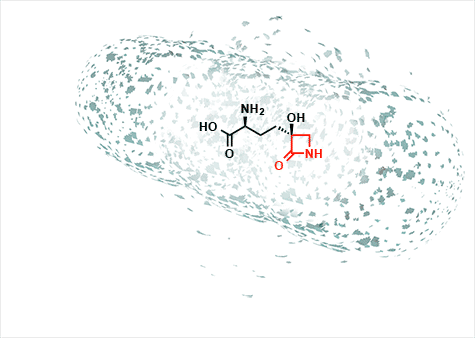
“In my lab we want to understand at a fundamental molecular level why this beta-lactam inhibits glutamine synthetase and not transpeptidases,” Wencewicz said. “This might open up a new chapter in the pharmacology of the enchanted ring.”
And there’s something else. Most antibiotics fail because they can’t cross the bacterial membrane, he said. Getting a molecule inside a bacterium is more difficult than getting it into a human cell because bacteria are surrounded by multi-layered, selectively permeable cell envelopes. One reason naturally occurring antibiotics such as tabtoxin are attractive to drug developers is that they have a built-in delivery system as well as a killing mechanism.
The high-throughput screening campaign launched by GlaxoSmithKline ultimately failed, Wencewicz said, because the libraries of chemical compounds they tested for antibacterial activity were designed to hit targets in cells like ours. The bacterial envelope is much harder to cross and most of the chemicals couldn’t get in the cells.
Tabtoxin has two parts, connected by a bond. One is a nontoxic amino acid, threonine, and the other is a toxic compound, tabtoxinine beta-lactam. The bacteria takes in the molecule thinking it is getting a free amino acid lunch, cleaves the bond to get access to the amino acid and inadvertently releases the potent toxin.
“You won’t find that type of fined tuned delivery system in a library of synthetic compounds sitting in a pharmaceutical company,” Wencewicz said. “Only nature engineers mechanisms like that.”
Wencewicz said tabtoxin might be useful against multidrug-resistant tuberculosis, because mycobacteria express glutamine synthetase on the outside of the cell and rely heavily on the enzyme to build their cell envelopes. “A group at UCLA has shown that inhibiting the enzyme shuts down TB in animal models,” he said, “and that has encouraged us.”
“I’m optimistic,” Wencewicz said. “There’s very little in the pipeline right now, but as clinical resistance rises and new antibiotic approvals decline, the problem will command more attention. I feel there will be an upswing in antibiotic discovery, and, through a coordinated effort by the government, companies, physicians, and universities, an outpouring of new drugs.”